Bupropion, an antidepressant medication introduced in the United States in 1989, has always stood apart from traditional antidepressants. Initially categorized as “atypical” due to its undefined neurotransmitter effects distinct from tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), and selective serotonin reuptake inhibitors (SSRIs), bupropion’s mechanism of action has become significantly clearer through extensive research over the last decade. This article delves into the intricate neuropharmacology of bupropion, explaining precisely How Bupropion Works to alleviate depression and examining how its unique mechanism translates to its clinical efficacy and tolerability profile.
While bupropion’s effectiveness in treating depression is comparable to that of SSRIs and TCAs, its pharmacological approach is fundamentally different. Unlike many antidepressants that primarily target the serotonin system, bupropion exhibits minimal serotonergic activity and does not directly interact with postsynaptic receptors. This distinctive characteristic underpins its favorable side effect profile, notably the absence of common antidepressant-associated issues like sexual dysfunction, weight gain, and sedation. Understanding how bupropion works at the neurochemical level is crucial for healthcare providers to make informed treatment decisions, tailoring pharmacotherapy to individual patient needs.
The Monoamine Hypothesis of Depression: A Shifting Paradigm
For decades, the monoamine hypothesis has been a cornerstone in understanding depression. This theory posits that depression arises from a deficiency in monoamine neurotransmitters – specifically serotonin, norepinephrine, and dopamine – within the brain. The initial evidence supporting this hypothesis was compelling: classical antidepressants were observed to boost monoamine function, while drugs that deplete monoamines, like reserpine, could induce depressive symptoms in vulnerable individuals. Furthermore, studies revealed that depressed patients often had lower levels of serotonin and norepinephrine metabolites in their cerebrospinal fluid and exhibited diminished neuroendocrine responses to monoamine agonists. Crucially, nearly all antidepressants available at the time were found to enhance some aspect of monoaminergic function.
However, contemporary perspectives on the neurobiology of depression view monoamine dysregulation not as the primary cause but rather as a contributing factor. Depression and the response to antidepressants are now believed to be mediated by complex, still not fully understood physiological pathways. Monoamines are considered to play an “upstream” role, influencing “downstream” events such as alterations in gene expression and protein synthesis, which are ultimately responsible for the manifestation of depression and the modulation of antidepressant responses.
Several observations challenge the idea of monoamines being the sole, primary cause of depression. Firstly, while antidepressants rapidly enhance monoamine levels at synapses within hours of administration, the therapeutic effects usually take days or weeks to emerge. This delay suggests that the downstream consequences of monoamine activation are more critical in the development of depression. Secondly, antidepressants, despite their comparable clinical efficacy, vary dramatically in their potency to affect monoamine systems, sometimes by a factor of over 1000. This disparity indicates that efficacy isn’t solely determined by the magnitude of monoamine enhancement. Thirdly, the diverse mechanisms by which antidepressants boost monoaminergic neurotransmission suggest that multiple monoamine pathways can converge on common downstream pathways relevant to depression. Finally, recent research highlights that antidepressants might increase levels of brain-derived neurotrophic factor (BDNF), a protein vital for neuronal health. This points towards a potential neuroprotective role of antidepressants, supported by findings that hippocampal neurogenesis might be necessary for their behavioral effects and that untreated chronic depression can lead to hippocampal volume loss in humans.
Unpacking the Neuropharmacology: How Bupropion Works on Neurotransmitters
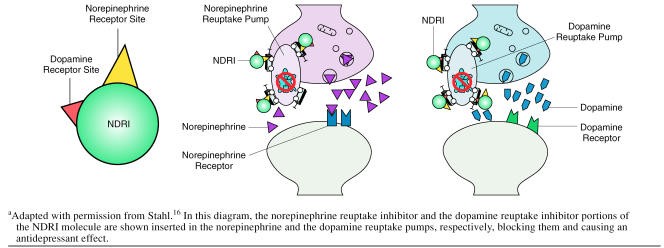
Animal studies have established that bupropion’s impact on monoaminergic neurotransmission is distinct from other antidepressants. Unlike many antidepressants, bupropion and its metabolites (hydroxybupropion, threo-hydrobupropion, and erythrohydrobupropion) do not directly alter serotonergic neurotransmission. They neither affect serotonin release or reuptake presynaptically nor bind to serotonin receptors postsynaptically. Instead, the primary mechanism of how bupropion works revolves around its interaction with dopamine and norepinephrine.
Bupropion and its major metabolite, hydroxybupropion, are potent inhibitors of dopamine and norepinephrine reuptake. They effectively reduce the reabsorption of these neurotransmitters back into presynaptic neurons in both rat and mouse studies. This reuptake inhibition leads to an increase in dopamine and norepinephrine concentrations in the synaptic cleft, the space between neurons where neurotransmitter signaling occurs. Consistent with this, acute administration of bupropion has been shown to reduce the firing rate of dopamine and norepinephrine neurons in rat brain stems in a dose-dependent manner. This reduction in firing is a feedback mechanism; increased synaptic neurotransmitter levels activate autoreceptors on the presynaptic neuron, signaling it to slow down neurotransmitter release.
Microdialysis studies, which directly measure neurotransmitter levels in living brains, further corroborate these findings. In mice models of depression, bupropion administration increased extracellular dopamine and norepinephrine concentrations specifically in the nucleus accumbens, a brain region associated with reward and motivation. Another microdialysis study in rats demonstrated increased dopamine and norepinephrine levels in the prefrontal cortex, an area crucial for executive functions and mood regulation. Furthermore, the importance of dopamine and norepinephrine reuptake inhibition for bupropion’s antidepressant effects is underscored by studies showing that blocking dopamine or norepinephrine receptors reduces the antidepressant efficacy of bupropion and its metabolite hydroxybupropion in animal models of depression. These preclinical data strongly indicate that how bupropion works as an antidepressant is primarily through its dual reuptake inhibition of dopamine and norepinephrine.
Image alt text: Diagram illustrating how a Norepinephrine-Dopamine Reuptake Inhibitor (NDRI) molecule, like bupropion, blocks both norepinephrine and dopamine reuptake pumps on a neuron, increasing the synaptic concentration of these neurotransmitters.
Clinical studies in humans reinforce the preclinical findings regarding how bupropion works. Studies using therapeutic doses of bupropion in depressed patients have demonstrated reduced whole-body norepinephrine turnover without altering plasma norepinephrine levels. This suggests that bupropion is indeed active in the central noradrenergic system. Moreover, positron emission tomography (PET) studies in healthy volunteers have shown that bupropion and its metabolites effectively bind to dopamine transporters in the striatum, a key brain region for dopamine activity, under steady-state conditions with typical therapeutic oral doses of bupropion SR (sustained-release). Dopamine transporter occupancy, a measure of how much bupropion is engaging with dopamine transporters, averaged around 26% and remained consistent even 24 hours after the last dose, indicating sustained action. These findings were supported by single photon emission computed tomography (SPECT) studies in depressed patients, which also reported similar dopamine transporter occupancy levels with therapeutic bupropion SR doses. While one study reported lower dopamine transporter occupancy, the lack of data variability and time course information makes interpretation challenging.
Further in vitro characterization of how bupropion works has been conducted using cells expressing human dopamine, norepinephrine, and serotonin transporters. These studies confirmed that bupropion and its metabolites inhibit reuptake at both human dopamine and norepinephrine transporters, with a slightly higher potency for dopamine transporters. Notably, serotonin reuptake inhibition was negligible, even at high concentrations. Brain concentrations of bupropion and its major metabolites remain above the 50% inhibitory concentrations (IC50) for dopamine and norepinephrine transporters throughout the typical 12-hour dosing interval of bupropion SR. This confirms that bupropion functions as a dual norepinephrine and dopamine reuptake inhibitor (NDRI) in humans at clinically relevant doses. Importantly, research has also shown that bupropion and its metabolites have minimal affinity for postsynaptic receptors, including histamine, adrenergic, serotonin, dopamine, or acetylcholine receptors. This lack of interaction with postsynaptic receptors is a key differentiator between bupropion and TCAs and some newer antidepressants that do interact with these receptors, contributing to different side effect profiles.
Image alt text: Graph depicting the in vivo binding of 11C-βCIT-FE, a dopamine transporter radioligand, at baseline and at 3, 12, and 24 hours after stopping steady-state dosing with bupropion SR, illustrating the sustained dopamine transporter occupancy by bupropion.
Image alt text: Bar graph comparing the combined relative in vitro potency (Cmax/IC50) of bupropion and its metabolites at human dopamine and norepinephrine transporters, showing the relative effectiveness of bupropion in inhibiting reuptake at these two transporters.
Collectively, the evidence clearly demonstrates that how bupropion works is by selectively inhibiting the reuptake of norepinephrine and dopamine in the human brain. It does not affect the release or transport of other neurotransmitters and does not bind to other neurotransmitter receptors to a significant extent. This unique pharmacological profile makes bupropion the only currently available NDRI that has been shown to increase dopamine neurotransmission in both the nucleus accumbens and the prefrontal cortex, brain regions critically implicated in mood regulation and reward pathways.
Clinical Efficacy and Applications: Translating Mechanism to Therapeutic Use
While the specific neurotransmitters targeted and the potency of neurotransmitter effects may differ, the overall effectiveness of antidepressant medications is generally comparable across different classes. This is supported by numerous studies and reflected in the guidelines of major psychiatric organizations. Despite its unique pharmacology, bupropion has consistently demonstrated antidepressant efficacy comparable to SSRIs and TCAs in head-to-head clinical trials. Pooled analyses of these trials even show identical remission rates between bupropion and SSRIs, highlighting that how bupropion works through a different pathway still achieves similar clinical outcomes in treating depression. Furthermore, bupropion has shown comparable efficacy when used in combination with the SSRI sertraline, even in patients with high baseline anxiety levels, suggesting its utility in treating depression with comorbid anxiety.
However, the distinctive neuropharmacological properties of bupropion, and specifically how bupropion works by targeting dopamine and norepinephrine, have significant clinical implications that extend beyond first-line antidepressant therapy. Bupropion is frequently used to augment the efficacy of serotonergic antidepressants, especially in cases of partial response to SSRIs or SNRIs. Moreover, its unique mechanism allows it to mitigate some of the common side effects associated with serotonergic antidepressants.
Beyond depression, bupropion’s mechanism of action makes it effective for other conditions characterized by dysfunctional noradrenergic and/or dopaminergic neurotransmission. Its dopamine reuptake inhibition properties are key to its effectiveness as a smoking cessation aid. By increasing dopamine levels, bupropion helps to reduce cravings and withdrawal symptoms associated with nicotine dependence. Clinical trials have consistently shown that bupropion approximately doubles short- and long-term smoking abstinence rates compared to placebo or nicotine patches. Bupropion is also effective in treating attention-deficit/hyperactivity disorder (ADHD), a condition thought to involve dysregulation of both noradrenergic and dopaminergic systems. It is also the only antidepressant proven effective in reducing the risk of seasonal depressive relapse when used prophylactically for seasonal affective disorder (SAD), further linking noradrenergic and dopaminergic pathways to the pathogenesis of both ADHD and SAD. Emerging data also suggest that bupropion may be less likely than TCAs to induce mania in bipolar depression, making it a preferred treatment option in this population. This lower risk of mania is hypothesized to be related to its lack of serotonergic properties and effects on postsynaptic β-receptors. In contrast to SSRIs, SNRIs, TCAs, and MAOIs, bupropion has not been extensively studied for anxiety disorders, reflecting its different neurochemical focus.
Clinical Tolerability: The Advantage of NDRI Mechanism
Unlike therapeutic effects that take weeks to manifest, most antidepressant side effects appear within hours or days of starting medication. This suggests a direct link between the acute synaptic effects of antidepressants and their tolerability profiles. The specific neurotransmitter effects of antidepressants indeed correlate with distinct side effect patterns. Because how bupropion works is unique among marketed antidepressants, its tolerability profile also stands out.
In placebo-controlled studies, the most common adverse events reported with bupropion were dry mouth, nausea, and insomnia, side effects also seen with other antidepressants. However, bupropion’s tolerability is distinguished by the absence of significantly increased rates of sexual dysfunction, weight gain, and sedation compared to placebo – side effects frequently associated with other classes of antidepressants.
The association of SSRIs, TCAs, MAOIs, and SNRIs with sexual dysfunction is well-documented. Studies have shown that sexual problems are significantly more likely with antidepressants affecting serotonergic function compared to bupropion, which consistently demonstrates the lowest risk of sexual dysfunction. Comparator trials between bupropion and SSRIs corroborate these findings. Bupropion has also been successfully used to switch patients off other antidepressants causing sexual dysfunction and has shown promise as an antidote for antidepressant-induced sexual side effects in numerous studies.
Weight gain is another common side effect with certain antidepressants, particularly SSRIs, potentially linked to serotonergic mechanisms and antihistaminergic effects. Bupropion, in contrast, is not associated with weight gain. Clinical trials suggest it is weight-neutral in patients of normal weight and may even lead to modest weight loss, especially in those with higher initial body mass index. Interestingly, bupropion has also shown efficacy as an adjunct for weight loss in non-depressed obese individuals, although the exact mechanism of this weight-reducing effect is still under investigation. Dopaminergic and noradrenergic pathways are known to play crucial roles in regulating appetite and satiety, potentially explaining this effect in line with how bupropion works.
Sedation is another side effect less likely with bupropion. Controlled clinical trials show no significant difference in sedation rates between bupropion SR and placebo. Pooled analyses comparing bupropion to SSRIs consistently demonstrate significantly lower rates of sedation with bupropion.
Seizure risk is an important consideration with antidepressants. While most newer antidepressants have a low seizure incidence (0.1% to 0.3%), TCAs have a higher rate. For bupropion, the reported seizure incidence is dose-dependent and remains relatively low, particularly with sustained-release formulations at recommended doses. The mechanisms by which antidepressants lower seizure threshold are not fully understood.
In summary, the dual norepinephrine and dopamine reuptake inhibition mechanism of how bupropion works results in a distinct side effect profile. While some common side effects like dry mouth, nausea, and insomnia may occur, bupropion stands out for its lower propensity to cause sexual dysfunction, weight gain, and sedation, offering a valuable alternative for patients concerned about these side effects.
CONCLUSIONS: Bupropion’s Unique Place in Antidepressant Therapy
Preclinical and clinical evidence unequivocally demonstrates that how bupropion works is through dual inhibition of norepinephrine and dopamine reuptake. This novel mechanism of antidepressant action translates into a unique clinical profile. Bupropion, as the only currently available NDRI, achieves antidepressant efficacy comparable to other classes of antidepressants without the burden of common antidepressant-associated side effects like sexual dysfunction, weight gain, and sedation. This favorable profile supports the use of bupropion as a first-line antidepressant and highlights its potential as a valuable augmentation therapy. Understanding how bupropion works empowers clinicians to leverage its distinct pharmacological properties to optimize treatment strategies and cater to the diverse needs of individuals struggling with depression and related conditions.
Drug names: amitriptyline (Elavil and others), bupropion (Wellbutrin, Zyban, and others), citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac and others), mirtazapine (Remeron), paroxetine (Paxil and others), phenelzine (Nardil), reserpine (Serpalan and others), sertraline (Zoloft), venlafaxine (Effexor).
Footnotes
Dr. Stahl has been a consultant for, received honoraria from, or conducted clinical research supported by Abbott, Asahi Kasei, AstraZeneca, Bristol-Myers Squibb, Cephalon, Cypress Bioscience, Eli Lilly, GlaxoSmithKline, Organon, Otsuka, Pfizer, Pierre Fabre, and Wyeth. Dr. Pradko has been a consultant for and has served on the speakers or advisory board of GlaxoSmithKline. Drs. Haight, Modell, Rockett, and Learned-Coughlin are employees of GlaxoSmithKline.